

Introduction
- Expressed sequence tags (ESTs) are short, single-pass sequence reads derived from cDNA libraries, generally 200-500 bp in length
- Represent portions of expressed genes; valuable for rapid gene discovery and genome annotation
- In 1983, SD Putney for the first time demonstrated the use of cDNA in identification of genome. The term expressed sequence tags (ESTs) was coined by Anthony Kerlavage at the Institute for Genomic Research. In 1991, Mark Adams used EST in relation to gene discovery and Human Genome project.
- *cDNA or complementary DNA is reversely transcribed from mRNA using reverse transcriptase enzyme. cDNA is widely used in cloning of genes in eukaryotes.
Contents
Expressed Sequence Tags (ESTs)
- Short sequences (200-500 nucleotides) randomly selected from a genome library.
- Used for identifying and mapping the entire genome of a species.
- Generated by sequencing the ends of DNA, providing cost-effective and fast genomic analysis.
- Can be used to search homologous organisms in databases like NCBI.
Generation of ESTs
- Isolation of mRNA from tissue/cells of interest
- Reverse transcription to generate complementary DNA (cDNA)
- More stable than mRNA
- Lacks intronic regions
- cDNA libraries constructed
- Random clones are selected and single-pass sequenced from one or both ends to derive 5′- and/or 3′-EST
- Typically automated Sanger sequencing used; next-generation platforms emerging for high-throughput EST analysis.
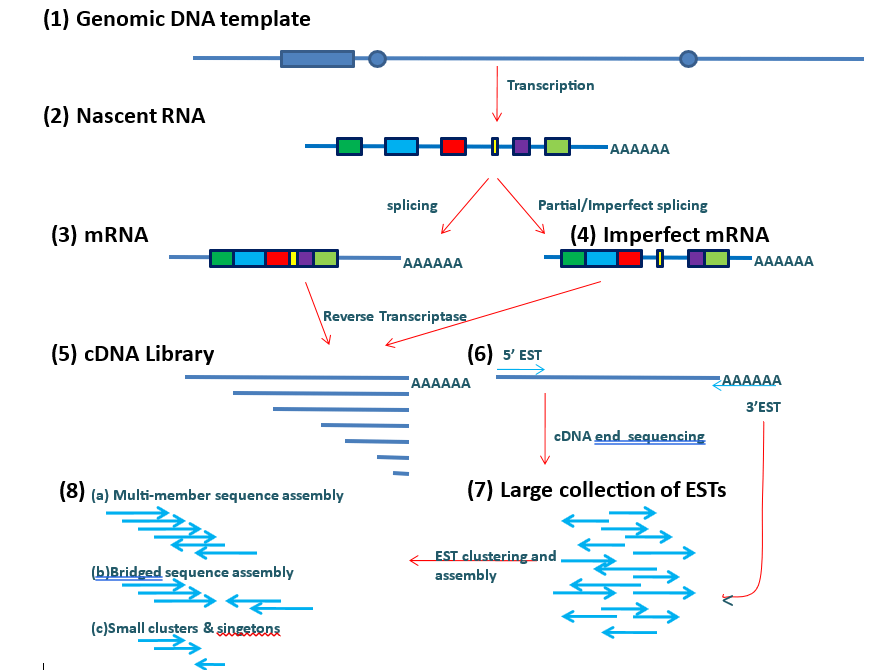
Method of construction of ESTs from nascent DNA. The process involves transcription of nascent DNA, reverse transcription of mRNA and finally EST synthesis and clustering.
Applications of ESTs
1. Gene discovery and genome annotation
- EST sequences can identify novel genes expressed in specific cell/tissue types.
- Matching to genomic sequence maps coding regions and refines genome annotation.
- Important resource for the human genome project; revealed >60% of human genes
2. Expression profiling
- The abundance of ESTs corresponding to a gene indicates its expression level.
- Differential EST analysis of normal vs diseased states informs on genes involved in pathogenesis
3. Alternative splicing analysis
- Alignment of ESTs to genomic sequence reveals different splice isoforms of genes
4. Evolutionary studies
- Orthologous genes in related species identified through EST sequence conservation
- Rates of divergence between species can be estimated
5. Contribution to the Human Genome Project
- Thousands of genes identified in the Human Genome Project based on ESTs. The Human Genome Project, initiated by the U.S. Department of Energy and the National Institutes of Health, completed in 2003.
Limitations and Future Directions
- Prone to sequencing errors leading to overestimation of gene numbers
- Incomplete coverage; rarely sample full transcript length
- Advances in RNA-sequencing now allow deep, quantitative transcriptome analysis
- But ESTs retain utility for poorly annotated genomes and gene mapping
Further learning resources
 |
 |
 |
Key References
- Adams et al. Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991.
- Nagaraj et al. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011.
- Varshney et al. Genetic molecular markers in plants: development and applications. Crit Rev Biotechnol. 2018.
- Zhao et al. Leveraging approaches from human genetics to identify causal genes and pathways in complex diseases. Front Genet. 2019.