

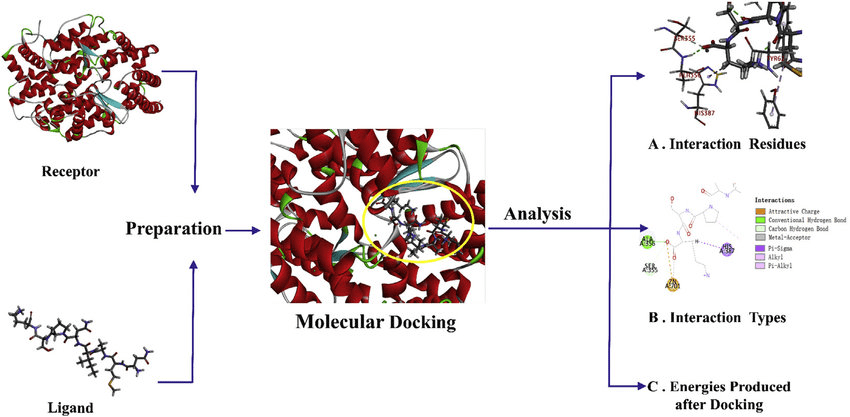
Molecular Docking and MD simulation
Molecular docking is a computational method used to predict the binding orientation and affinity between a small molecule (ligand) and a target protein (receptor). Molecular docking is a key tool in structural molecular biology and computer-assisted drug design. The goal of ligand—protein docking is to predict the predominant binding mode(s) of a ligand with a protein of known three-dimensional structure.
Docking can be used to perform virtual screening on large libraries of compounds, rank the results, and propose structural hypotheses of how the ligands inhibit the target, which is invaluable in lead optimization
Binding energy is calculated as a quantitative measure of the affinity between the ligand and the protein. It takes into account various contributing factors, such as electrostatic interactions, van der Waals forces, hydrogen bonding, hydrophobic interactions, and desolvation effects.
Docking Basics
- Initially – Receptor (protein) and ligand rigid
- Most current approaches – Receptor rigid, ligand flexible
- Advanced approaches – Receptor (to a degree) and ligand flexible
Stages of Docking
Pose Generation
- Rigid docking with a series of conformers: Most techniques use this approach and Most techniques will generate the conformers internally rather than using conformers as inputs.
- Incremental construction (FlexX): Split ligand into base fragment and side-chains, Place base and Add sidechains to grow, scoring as you grow
- In general, use a very basic vdW shape function
- Often see variability with input conformers
Pose Selection/Scoring
- Where most of the current research focused
- More sophisticated scoring functions take longer:
- Balance need for speed vs. need for accuracy
- Virtual screening needs to be very fast
- Studies on single compounds can be much slower
- Can do multi-stage studies
Receptor Preparation
- Dependent on docking program used
- Structure selection
- Site selection
- Add charges
- Often have to add hydrogens, some programs more sensitive to positions than other
- Remove/include waters, cofactors, metals
- Pre-docking refinement
- Remember to consider missing residues or atoms
Ligand preparation
- Input structures (extract from PDB, draw, convert from SMILES)
- Add bond orders
- Generate isomers if chiral centers
- Calculate charges
– Predict pKa’s for each potential charged atom
– Generate a structure for each charge combination for a given pH range (e.g., 5-9) - Minimize structures – Generally using a molecular mechanics forcefield
- For Screening, can download public sets from
ZINC (available compounds) or PubChem
Applications of Molecular Docking
- It can demonstrate the feasibility of any task, if it carried out before experimental part of any investigation
- interaction investigations between small molecules (ligand) and protein target (may be an enzyme) may predict the activation or inhibition of enzyme. Such type of information may provide a raw material for the rational drug designing.
- Molecular docking can predict an optimized orientation of small molecule or ligand on its target. It can predict different binding modes of ligand in the groove of target molecule.
- Molecular docking in combination with scoring function can be used to screen huge databases for finding out potent drug candidates in silico, which can target the molecule of interest
Docking Packages
- Free
– AutoDock (Art Olsen, David Goodsell, Scripps)
– UCSF DOCK (Kuntz Group) - Commercial
– Glide (Schrodinger)
– GOLD (CCDC)
– FlexX (BiosolveIT)
– ICM (Molsoft)
– Surflex (Tripos
AutoDock
AutoDock is a popular software package commonly used for molecular docking studies.
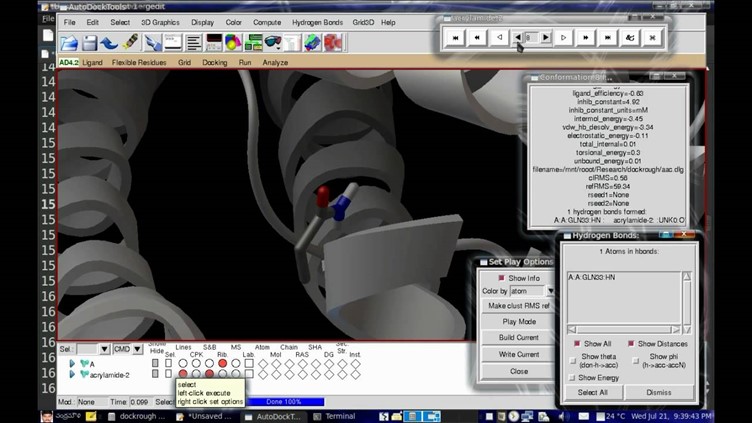
Steps involved in molecular docking using AutoDock
- Preparation of the target protein
- Obtain the three-dimensional structure of the target protein in a suitable file format, such as PDB (Protein Data Bank) format.
- Remove any water molecules, cofactors, or other ligands not involved in the binding site.
- Assign Gasteiger charges to the protein atoms.
- Save the prepared protein file in PDB format.
- Preparation of the ligand molecule:
- Obtain the three-dimensional structure of the ligand in a suitable file format, such as PDB or MOL2 (Tripos Mol2) format.
- Assign Gasteiger charges or other appropriate charges to the ligand atoms.
- Save the prepared ligand file in PDB or MOL2 format.
- Setting up the AutoGrid parameter file
- Launch the AutoGrid program included with AutoDock.
- Define the grid box parameters to define the search space around the binding site of the target protein.
- Specify the spacing and dimensions of the grid, which should be large enough to accommodate the ligand.
- Save the AutoGrid parameter file (GPF) for later use in docking.
- Setting up the AutoDock parameter file
- Launch the AutoDock program.
- Load the prepared protein file and ligand file.
- Set up docking parameters, such as the number of genetic algorithm runs and population size.
- Specify the location and name of the output files.
- Save the AutoDock parameter file (DPF) for later use in docking.
- Running the AutoGrid program.
- Execute the AutoGrid program using the saved AutoGrid parameter file (GPF).
- The program will generate a grid map file (GPF) that describes the protein’s interaction potentials.
- Running the AutoDock program.
- Execute the AutoDock program using the saved AutoDock parameter file (DPF).
- AutoDock will perform the docking calculations using the Lamarckian Genetic Algorithm (LGA) or other chosen algorithms.
- The program will generate multiple docking conformations or poses of the ligand within the defined search space.
- Analyzing the results
- Examine the output files generated by AutoDock, such as the docking log file (DLG) or binding energy file.
- Identify the top-ranked docking conformations based on their binding affinity or energy scores.
- Visualize the protein-ligand complexes to analyze the binding interactions and make further interpretations.
MD Simulation
Molecular dynamics (MD) simulation is a computational technique used to study the behavior of atoms and molecules over time.
- Force fields typically include terms for bonded interactions (e.g., bond stretching, angle bending), non-bonded interactions (e.g., van der Waals forces, electrostatic interactions), and solvent effects.
- MD Simulations has been used to study protein function, protein stability, protein-protein interaction, enzymatic reactions and drug-protein interactions, and membrane proteins
Reference
- Paul Sanschagrin (bu.edu)
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384-13421. Published 2015 Jul 22. doi:10.3390/molecules200713384
- Pinzi L, Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. International Journal of Molecular Sciences. 2019; 20(18):4331. https://doi.org/10.3390/ijms20184331
- Tu, Maolin & Cheng, Shuzhen & Lu, Weihong & Du, Ming. (2018). Advancement and prospects of bioinformatics analysis for studying bioactive peptides from food-derived protein: Sequence, structure, and functions. TrAC Trends in Analytical Chemistry. 105. 10.1016/j.trac.2018.04.005.