


Introduction
Michael Smith, a British-born Canadian biochemist won the 1993 Nobel Prize (with Kary B. Mullis) in Chemistry for the development of a technique called – oligonucleotide-based site-directed mutagenesis, which enabled researchers to introduce specific mutations into genes and, thus, to the proteins that they encode. Site-directed mutagenesis (SDM) is a method to create specific, targeted changes in Double stranded plasmid DNA.
Reasons to make specific DNA alteration
- To study changes in protein activity that occur as a result of the
DNA manipulation. - To select or screen for mutations (at the DNA, RNA or protein
level) that have a desired property. - To introduce or remove restriction endonuclease sites or tags.
Strategies of directed mutagenesis
- Oligonucleotide-Directed Mutagenesis with M13 DNA
- Oligonucleotide-Directed Mutagenesis with Plasmid DNA
- PCR based mutagenesis
- PCR-Amplified Oligonucleotide-Directed Mutagenesis
- Error-Prone PCR
- Random Mutagenesis with Degenerate Oligonucleotide Primers
- Random Insertion/Deletion Mutagenesis
- DNA Shuffling
- Mutant Proteins with Unusual Amino Acids
Oligonucleotide-directed mutagenesis with M13 DNA
- Single-stranded bacteriophage M13 (M13 + strand), carrying a cloned gene is taken.
- It is annealed with a complementary synthetic oligonucleotide containing one mismatched base.
- With the oligonucleotide as the primer, DNA synthesis is catalyzed by the Klenow fragment of E. coli DNA polymerase I.
- Synthesis continues until the entire strand is copied. The newly synthesized DNA strand is circularized by T4 DNA ligase.
- The ligation reaction mixture is used to transform E. coli.
- Both the target DNA with its original sequence and the mutated sequence are present in the progeny M13 phage.
- After the double-stranded form of M13 is isolated, the mutated gene is excised by digestion with restriction enzymes and then spliced onto an E. coli plasmid expression vector.
- For further study, the altered protein is expressed in and purified from the E. coli cells.
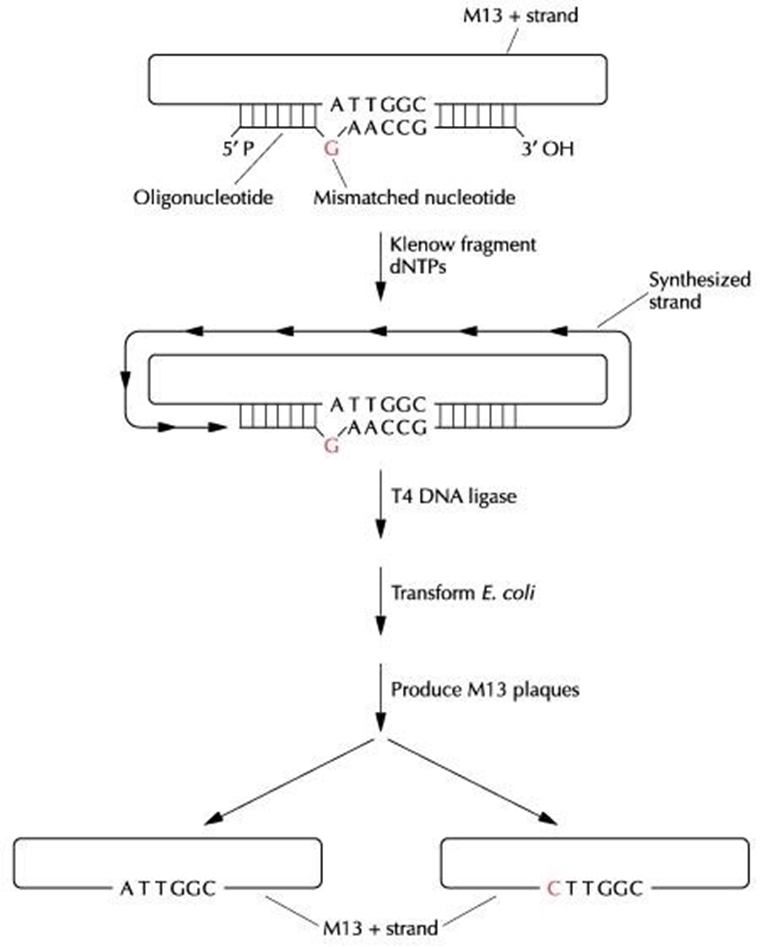
- As per the diagram we see only 50% of the M13 viruses are expected to carrying the mutated form of the target.
- In practice only around 1% of the plaques actually contain phage carrying the mutated gene.
- Consequently, the oligonucleotide-directed mutagenesis method has been modified in several ways to enrich for the number of mutant phage plaques that can be obtained
Modification of Oligonucleotide-directed mutagenesis with
M13 DNA
Introduce the M13 viral vector carrying the gene that is to be mutagenized into an E. coli strain that has two defective enzymes of DNA metabolism
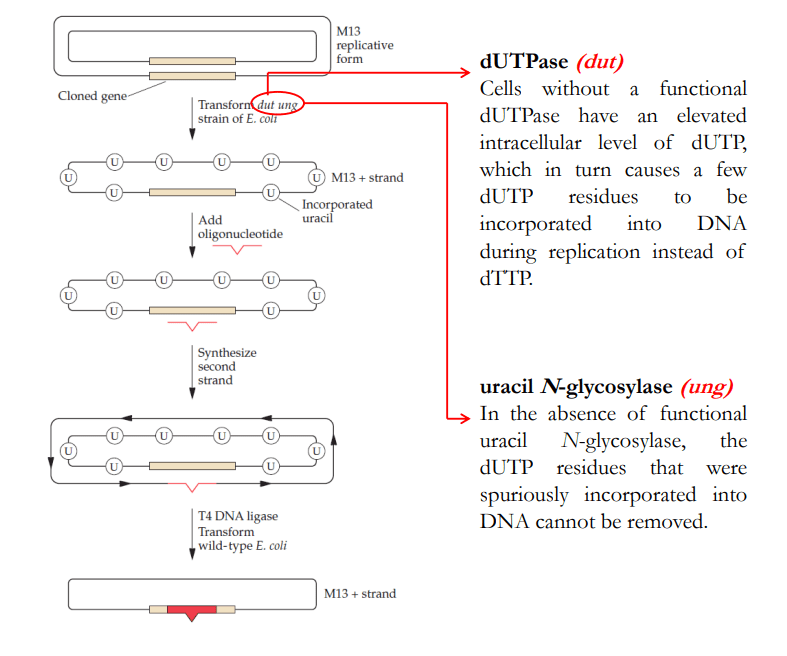
- The target DNA is cloned into the double-stranded replicative form ofbacteriophage M13, which is then used to transform a dut ung strain of E. coli.
- The dut mutation causes the intracellular level of dUTP to be elevated; the high level of nucleotide leads to the incorporation of a few dUTP residues (U). The ung mutation prevents the removal of any incorporated uracil residues.
- Following in vitro oligonucleotide-directed mutagenesis, the double stranded M13 vector with the mutated DNA is introduced into wild-typeE. coli.
- The wild-type ung gene product removes any uracil residues from the parental strand, so a significant portion of the parental strand is degraded.
- The mutated strand remains intact because it does not contain uracil. It serves as a template for DNA replication, thereby enriching the yield of M13 bacteriophage carrying the mutated gene
- Efficiency of mutant production is in the range of 10–45% and no precautions to prevent mismatch repair are required.1
Drawbacks of using the M13 DNA
- There is a need to subclone a target gene from a plasmid into M13 and then, after mutagenesis, clone it back into a plasmid.
- Additional step of transforming enzyme defective E. coli necessary to enrich the yield.
- Lengthy process involving multiple steps
Oligonucleotide-Directed Mutagenesis with Plasmid DNA
- The target DNA is inserted into the multiple cloning site (MCS) on the vector pALTER.
- Plasmid DNA is isolated from E. coli cells and alkaline denatured.
- Mutagenic oligonucleotide, the ampicillin resistance (Ampr) oligonucleotide, and the tetracycline sensitivity (Tets) oligonucleotide are annealed.
- The oligonucleotides act as primers for DNA synthesis by T4 DNA polymerase with the original strand as the template.
- The gaps between the synthesized pieces of DNA are sealed by T4 DNA ligase.
- The reaction mixture is used to transform E. coli host cells, and cells that are Ampr and Tets are selected
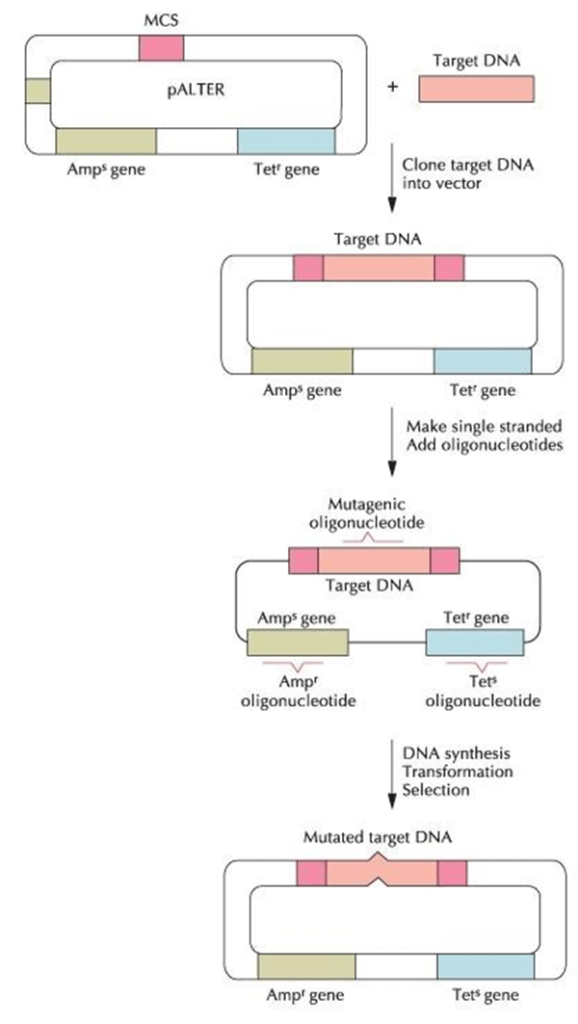
- With this procedure, about 90% of the selected transformants have the specified mutation in the target gene.
- In the remaining transformants, the target gene is unchanged because the oligonucleotide did not anneal to the target gene or it was bypassed during DNA synthesis.
- The cells with the specified mutation in the target gene are identified by DNA hybridization.
- All of the plasmids, host bacterial strains, enzymes, oligonucleotides (other than the one needed to alter the target gene), and buffers for this method are sold as a kit, facilitating its widespread use
Cassette mutagenesis
Cassette mutagenesis provides a powerful and versatile approach for the generation of mutations and allows a wide range of studies of the relationship between base sequence and function in nucleic acids or between structure and function in proteins. The central aspect of this approach is the replacement of a region of DNA with new sequences that can be of any length, any sequence or any mixture of sequences. Thus, in studying the basis for protein function one can introduce not only changes of a few amino acids (an important aspect of primer-extension in vitro mutagenesis) but can also create chimeric proteins in which entire domains of a protein have been replaced with a region of totally different amino acid sequence.2
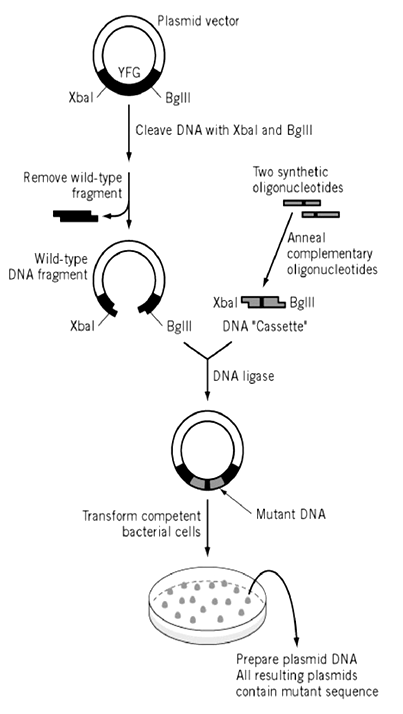
This method can generate mutants at close to 100% efficiency, but in practice limited by the availability of suitable restriction sites.
References
- Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21054/
- GEB, Sem VI, B.Sc (H) Biochemistry Dr. Jayita Thakur
- Mark J. Zoller, Michael Smith, Oligonucleotide-directed mutagenesis using M13-derived vectors: an efficient and general procedure for the production of point mutations in any fragment of DNA, Nucleic Acids Research, Volume 10, Issue 20, 25 October 1982, Pages 6487–6500, ↩︎
- Richards, John H, ‘Cassette mutagenesis’, in M J McPherson (ed.), Directed Mutagenesis: A Practical Approach (Oxford, 1991; online edition, Oxford Academic, 31 Oct.2023), accessed 9 June 2024 ↩︎